Ohne Biologiestudium ist es eine ziemliche Herausforderung die Ansätze in der Forschung zu verstehen.
Durch das Anfang 2022 erschienene Buch „Das Ende aller Leiden“ bin ich ein ganzes Stück klüger geworden. Ich habe versucht die für mich in Bezug auf die Erforschung von Therapien für Muskelkranke interessantesten Aspekte zusammenzufassen und auch mit Bildern verständlicher zu machen.

In dem brandaktuellen Buch „Das Ende aller Leiden“ mit dem Untertitel „Wie RNA-Therapien die Behandlung von Krebs, Herzkrankheiten und Infektionen revolutionieren“ beschreiben die beiden Wissenschaftsjournalisten Edda Grabar und Ulrich Bahnsen anschaulich die Geschichte der Entdeckung von RNA als Instrument zur Bekämpfung von unterschiedlichen Krankheiten. Corona hat die Forschung rund um die RNA-Medizin kräftig verstärkt, sodass sie nun in den Fokus rückt, schwere Leiden wie Krebs und Erbkrankheiten, aber auch Infektionen wie HIV, Tuberkulose oder Malarie zu therapieren.
RNA kann, so wie wir das von einigen Corona-Impfungen kennen, den Immunzellen zeigen, wie ein gefährlicher Erreger aussieht, sodass das Immunsystem ihn schneller erkennen und bekämpfen kann.
RNA kann als Bote wichtige Instrumente zum Reparieren von defekten Genen liefern und sogar selbst fehlerhafte Gene im laufenden Betrieb reparieren.
Um zu verstehen wo die Therapien für Muskelkranke ansetzen, muss man zunächst ungefähr verstehen, worin die Ursache der Erkrankung begründet ist.
In den rund 21.000 menschlichen Genen konnten Forschende mehr als 5.000 krankheitsverursachende Veränderungen (Mutationen) entdecken. Eine Veränderung in der Basenabfolge des Genoms bedeutet oft eine Störung in der Protein- oder Enzymherstellung, die für einen gesunden Mechanismus im Körper wichtig sein.
1. Wie kann der Körper eigentlich auf der Grundlage von bestimmten Basenabfolgen z.B. Proteine herstellen und welche Rolle spielt die RNA dabei?
Die DNA des Menschen besteht aus der Anordnung von Basenpaaren.
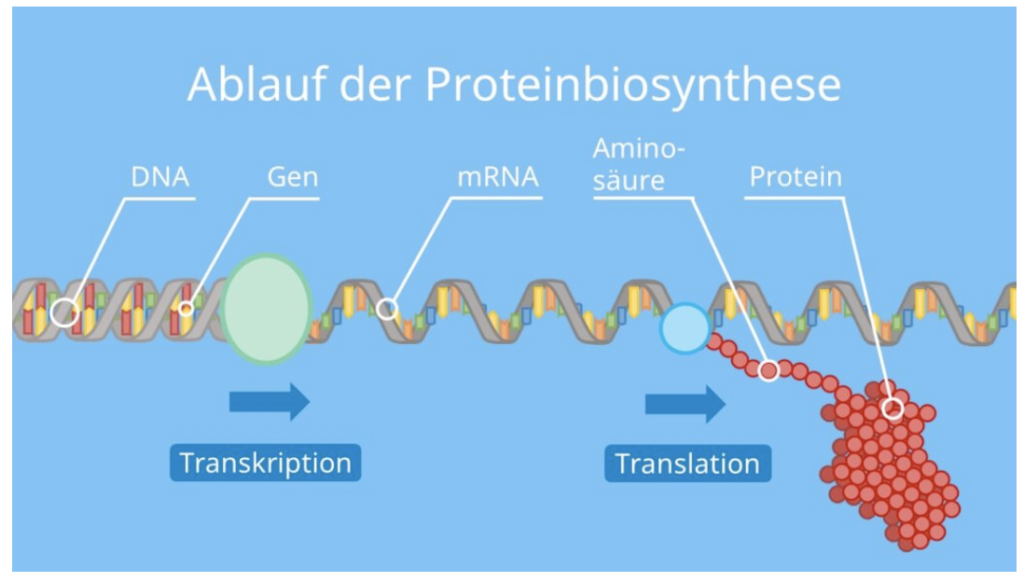
Die Anweisung für die Herstellung von Proteinen ist in DNA-Sequenzen kodiert. Das heißt, eine bestimmte Abfolge der Basenpaare führt zu der Herstellung von bestimmten Proteinen (oder anderen Molekülen). Dafür wird die DNA „abgelesen“.
Der Prozess des „Lesens“ von DNA-Sequenzen erfordert zwei Phasen: Transkription und Translation. Bei der Transkription wird die Erbinformation der DNA in eine mRNA transkribiert, also umgeschrieben.
Die mRNA ist die Boten-RNA (auch guide RNA), die die Informationen den Ribosomen in der Zelle überbringt. So muss die DNA nicht transportiert werden und sie wird auch nicht beschädigt!
In den Ribosomen (Proteinfabrik der Zelle) kann dann die Translation ablaufen. So wird Aminosöure für Aminosäure angehängt, bis das gesamte Gen in das jeweilige Protein übersetzt ist.
2. Was bedeutet „Spleißen“?
Die mRNA ist nicht die simple Kopie der DNA. Sie wird durch die mRNA weiterbearbeitet und modifiziert. Das nennt man „spleißen“.
Durch das Spleißen können aus demselben Gen verschiedene Gewebe entstehen. So erklärt sich auch, dass aus ca. 21.000 menschlichen Genen an die 1 Million Proteinspezies entstehen können.

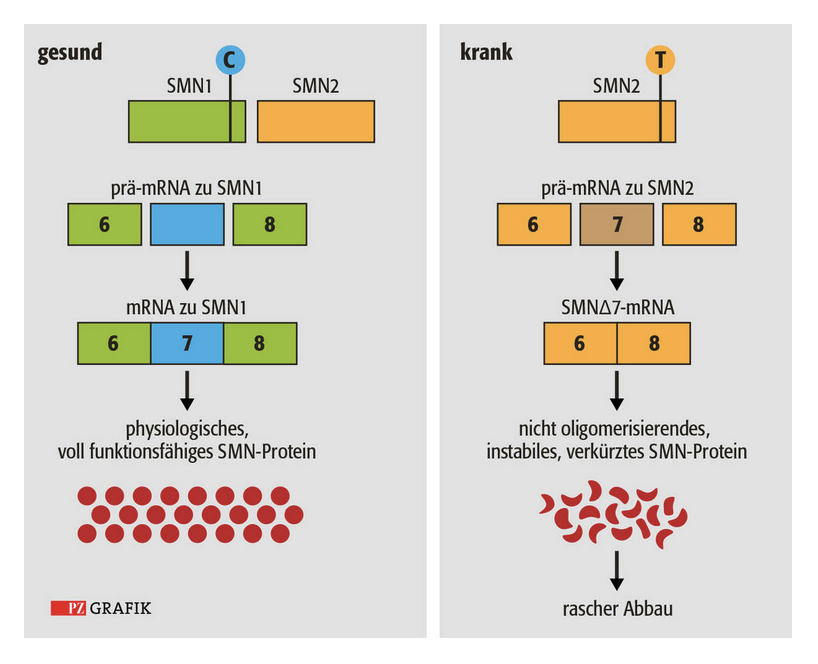
Klar, dass beim Spleißen auch Fehler entstehen. Diese haben meist keine gravierenden Auswirkungen. Aber manchmal eben doch. So wie bei der spinalen Muskelatrophie (SMA), eine der schwerwiegendsten Muskelerkrankungen.
Bei der SMA wird aufgrund eines genetischen Defekts nicht genug SMN-Protein (Survival of Motor Neuron) gebildet. An dieser Stelle setzt das Antisensenukleotid Nusinersen (Spinraza®) an. Der Wirkstoff verändert, wie die prä-mRNA von SMN2 gespleißt wird, was letztlich dazu führt, dass vollständiges und funktionsfähiges SMN-Protein in größeren Mengen gebildet werden kann.
Das Therapeutikum greift also in den Spleißvorgang ein und ist somit eine RNA basierte Therapie.
3. Was ist der Unterschied zwischen einer RNA-Therapie und einer Gentherapie?
Spinraza® ist das erste Medikament gegen SMA. Es wurde 2016 von der FDA (dem amerikanischen Paul Ehrlich Institut) zugelassen.
Es liefert den Beweis für das Prinzip: man kann RNA Fehler therapieren; das fehlerhafte Spleißen der mRNA wird korrigiert.
Zolgensma hingegen, das zweite Medikament im Kampf gegen SMA (von AveXis entwickelt) setzt an der der DNA an.
Mithilfe eines abgeschwächten Virus wird das Medikament als DNA in alle Körperzellen geschleust. So wird das gesamte fehlerhafte SMN1-Gen ersetzt. Somit ist dies eine Gentherapie.
Es können nur Kinder nehmen, dessen Abwehrsystem noch nicht gelernt hat gegen jenes Virus vorzugehen, mit dessen Hilfe das SMN1-Gen in die Zelle transportiert wird.
4. Wie werden die Erkenntnisse nun für die Therapie von LMNA-basierten Mutationen genutzt?
Majes Defekt zählt zu den „seltene Erkrankung“. Gut 80 % aller seltenen Erkrankungen beruhen auf einem Genfehler. Je mehr Forschung zu einzelnen Gendeffekte passiert, um so mehr Menschen mit „seltenen Erkrankungen“ kann man zukünftig eine Therapie anbieten. Denn die Erkenntnisse lassen sich übertragen und bringen so Nutzen für eine sehr große Zielgruppe, zu der auch Ich und Du irgendwann zählen könnte.
Majes Symptome sind auf eine Mutation auf dem LAMINgen zurückzuführen. Das Gen, was Lamin exprimiert liegt auf dem ersten Chromosom. Dort haben die Genetiker einen Fehler entdeckt: Da wo die Base „Adenin“ liegen müsste, sitzt bei Maje „Guanin“. Die sogenannte Punktmutation oder „missense mutation“ hindert die Zelle daran das Protein Lamin in ausreichender Dosis herzustellen, was für die Stabilität der Zellwand aber wichtig ist. Wird der menschliche Muskel nicht mit genügend Lamin versorgt, kann die Muskulatur nur bedingt wachsen.
Der Forschungsansatz für Majes Mutation beruht darauf, das G durch ein A zu ersetzen. Die Methode nennt man „base editing“, weil die Base bearbeitet wird.
Base Editors sind veränderte Genscheren, die aber die DNA nicht durchschneiden sondern einen Zuckerrest „abknipsen“, so dass eine einzelne Base ausgetauscht werden kann. Weil der DNA-Schnitt vermieden wird, wird das Gen-Editing mit einem Base Editor generell als sicherer angesehen als mit der klassischen Genschere.
An Majes iPS Zellen hat die Korrektur mithilfe des base editors „SpRY-CBE“ erfolgreich funktioniert.
Bei den Experimenten an ihren primären Muskelzellen hat sich gezeigt, dass die Genpincette nicht nur dort schneidet, wo sie schneiden soll, sondern auch an anderen Stellen. Das nennt man „off-target Effekte“. Diese Nebeneffekte sind zum gewissen Grad normal und oft haben sie keine Auswirkungen auf den Organismus. In einigen Fällen können sie aber zu anderen Krankheiten führen. Womit das Problem nicht behoben würde, sondern nur verlagert.
Nun wurde zum Glück ein neuer base editor entwickelt, der sich noch besser für Majes Mutation eignet. Diesen möchte das Team ausprobieren.

Gelingt die Genkorrektur, würden Maje die korrigierten Zellen in den Muskel zurückgegeben werden, was ihre Muskelkraft verstärken soll.

