Was bedeutet das für Maje?
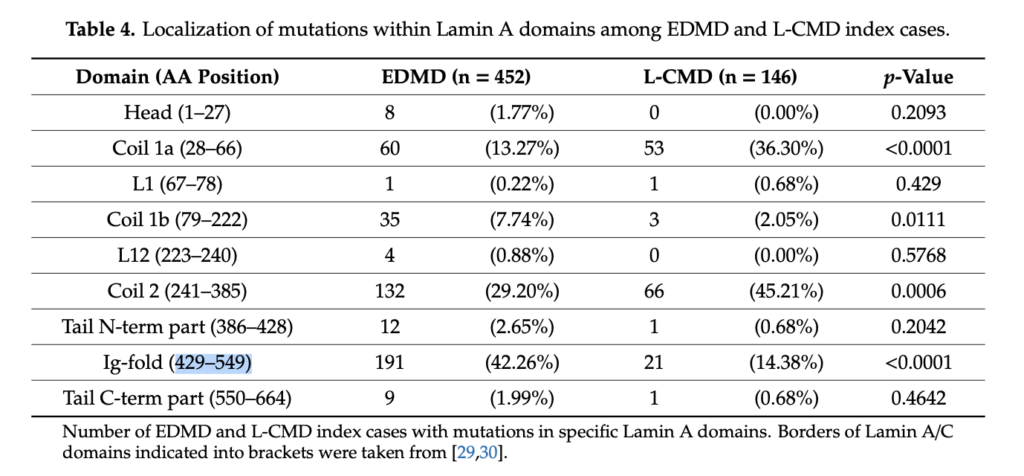
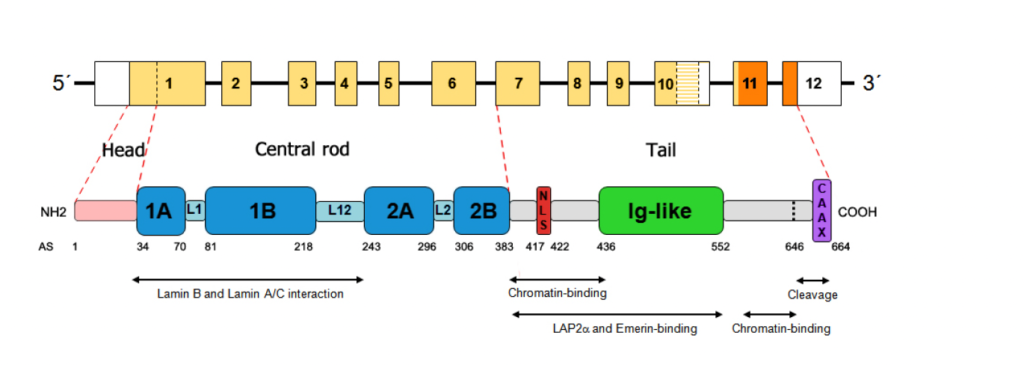
Majes Mutation befindet sich auf der Eiweißebene an der Stelle Asn456Asp (c.1366 A>G). Das heißt, dass sie mit höherer Wahrscheinlichkeit einen eher milderen Verlauf haben wird, aber mit einer 10% Wahrscheinlichkeit einen eher gravierenderen Verlauf.
Majes Symptomatik ist weder mild noch gravierend. Sie ist irgendwas dazwischen.
Die Krankheit tritt bei ihr erstmals innerhalb der Familie auf (Neumutation). Ihre Muskeln waren von Geburt an nicht gesund ausgeprägt, weil das Lamin nicht korrekt und ausreichend produziert wird. Dadurch bauen sich ihre Muskeln im Körper schneller ab und zerfallen. Mit der Zeit wird Maje immer schwächer. Die meisten Hauptmuskelgruppen des Körpers sind vom Abbau betroffen und im Laufe der Zeit auch das Herz und die Lunge.
Maje ist jetzt 9 und auf einen Elektrorollstuhl angewiesen. Während sie bis zu ihrem 6. Lebensjahr noch sicher laufen und Fahrradfahren konnte, schafft sie nun nur noch ein paar Meter in der Wohnung. Sie ist insgesamt sehr schwach. Es fällt ihr schwer vom Boden aufzustehen, frei zu sitzen oder ein Glas zum Mund zu heben.
Aufgrund der schwachen Rückenmuskulatur hat sie eine starke Wirbelsäulenverkrümmung entwickelt und trägt täglich ein Korsett, was die operative Versteifung ihrer Wirbelsäule hinauszögern soll. Überhaupt geht sie so selbstverständlich mit ihrer Situation um. Sie klagt kaum, hat zum Glück keine Schmerzen und ist insgesamt ein sehr fröhliches und unbeschwertes Kind.
Leider gibt es derzeit keine Heilung oder Behandlung, die den Prozess der Krankheit verlangsamen könnte. Das wollen wir ändern!